Thèse présentée à la Faculté
des sciences de l'Université de Paris
pour obtenir le grade de
Docteur es Sciences physiques
par
Hugh
Felkin
Etude
du mécanisme de la désamination nitreuse et des transpositions rétropinacolique
et pinacolique
soutenue le 19 juin 1954
devant la commission
d'examen :
Bauer, E., président
Prévost, C., Normant, H., examinateurs
INTRODUCTION
L'exposé du présent travail est divisé en deux parties
(1°) La partie générale, qui comporte cinq chapitres.
Après un bref aperçu historique sur la réaction de désamination nitreuse et les
transpositions rétropinacolique et pinacolique (chap. l), nous exposons les
théories actuelles concernant la réaction de désamination nitreuse (chap. Il);
les chapitres III.et IV sont consacrés à une discussion des résultats de nos
propres recherches en ce qui concerne le mécanisme de la désamination nitreuse
d'une part, celui des transpositions rétropinacolique et pinacolique d'autre
part. Enfin, le chapitre V est consacré à l'exposition générale des méthodes
expérimentales employées au cours de ce travail et des résultats obtenus,
(2°) La partie expérimentale, qui comprend la description
détaillée des modes opératoires employés et des composés obtenus.
Le but de la présente introduction est d'une part de
résumer les différents chapitres de la partie générale et de faciliter la
lecture de ces chapitres pris isolément, et d'autre part de dégager et de
grouper les conclusions expérimentales et théoriques qui découlent de nos
recherches personnelles.
Historique.- La réaction de désamination nitreuse des composés aminés
aliphatiques primaires est connue depuis plus d'un siècle. Les nombreux travaux
qui lui ont été consacrés depuis sa découverte par Piria en 1846 ont montré
qu'il s'agit d'une réaction extrêmement générale ; toutes les amines
aliphatiques primaires, sans exception, la subissent dans des conditions
appropriées.
Cependant, malgré sa grande généralité, la désamination
nitreuse se trouve parmi les réactions de chimie organique qui possèdent le
moins d'applications pratiques ; elle n'est guère employée que dans le
dosage des composés aminés primaires (van Slyke) et dans l'élégante méthode
d'extension de cycles carbonés de Tiffeneau et Tchoubar.
Ce manque d'applications pratiques est dû essentiellement
au fait que la désamination nitreuse est une réaction beaucoup plus complexe
que ne le laisse supposer l'équation
R-NH2 +
HNO2 ———— R-OH + N2
par laquelle elle est souvent représentée. En réalité, la
réaction conduit généralement à un mélange de -produits et le composé hydroxylé
R-OH attendu est parfois totalement absent. Dans le cas particulier des aminés
simples, le mélange obtenu peut. être constitué par différents composés du type
R-Y (Y = OH, Cl, OCOCH3, etc.) accompagnés de composés éthyléniques et de
composés (R'-Y et oléfines) résultant d'une transposition moléculaire et
possédant une structure différente de celle du composé aminé initial.
Entre 1850 et 1930 environ, une masse considérable de
faits expérimentaux s'est accumulée concernant la réaction de désamination
nitreuse et les transpositions du type rétropinacolique (aminés simples) ou
pinacolique (a-amino-alcools)
qui l'accompagnent souvent. C'est ainsi qu'ont été mises en évidence tout
d'abord l'influence qu'exerce la structure du composé aminé étudié sur la
nature des produits de la. réaction et ensuite, dans de nombreux cas,
l'influence non moins importante des conditions expérimentales sur la nature de
ces produits.
Cependant, les premières théories qui ont été émises pour
expliquer ces faits expérimentaux sont toutes peu satisfaisantes et le
mécanisme, aussi bien de la désamination nitreuse que des transpositions
rétropinacolique et pinacolique, est resté obscur pendant très longtemps.
Théories actuelles sur le mécanisme de la désamination
nitreuse.- Grâce à la
théorie électronique de la valence (Lewis) et aux travaux qui s'en sont
inspirés (Hückol, Ingold, Meerwein, Prévost, Robinson, Whitmore), de grands
progrès ont été réalisés au cours des trente dernières années dans
l'interprétation des mécanismes réactionnels. II est apparu en particulier que
les composés intermédiaires formés au cours do réactions complexes ne sont pas
nécessairement constitués par des corps stables et isolables, mais sont souvent
des composés dont la réactivité est telle qu'ils sont impossibles à isoler dans
les conditions mêmes de leur formation.
D'autre part, ces travaux ont montré qu'il existe un
nombre restreint de mécanismes réactionnels fondamentaux et que ces mécanismes
pouvant être caractérisés par différentes méthodes qui sont complémentaires.
Ces méthodes sont : (l°) l'étude de la cinétique de la réaction; (2°) l'étude
de l'influence qu'exerce le milieu réactionnel sur la nature des produits
obtenus; (3°) l'étude de l'influence qu'exerce la structure du composé étudié
sur la nature des produits de la réaction ; et (4°) l'étude des changements de
configuration stérique que subissent les composés optiquement actifs au cours
de la réaction.
L'application de ces différents critères à la
désamination nitreuse a conduit à la conclusion, généralement admise
aujourd'hui, que les premiers stades de la réaction sont en tous points
analogues à la réaction de diazotation en série aromatique
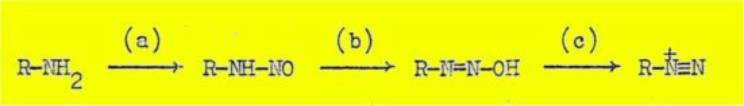
et que les derniers stades comportent la décomposition de
l'ion diazonium aliphatique par un mécanisme du type monomoléculaire, avec
formation intermédiaire d'un ion carbonium :
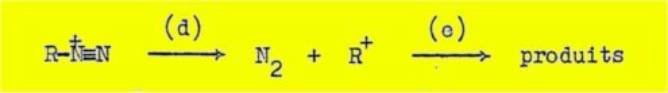
Les données qui étayent cette interprétation du mécanisme
de la désamination nitreuse sont les suivantes : stade (a) - cinétique ; stades
(b) et (c) - différents raisonnements analogiques ; stades (d) et (e) -
influence de la structure du composé aminé et des conditions expérimentales sur
la nature des produits obtenus et conséquences stériques de la réaction lorsque
celle-ci est appliquée à des composés optiquement actifs. Ces différents
arguments, qui ne semblent pas avoir été rassemblés auparavant, sont exposés en
détail dans le chapitre II.
Théories actuelles sur le mécanisme des transpositions
rétropinacolique et pinacolique.- Depuis les travaux classiques de Meerwein, effectués il y a
trente ans, il est admis que ces transpositions se font avec formation
intermédiaire d'un ion carbonium. Ce fait constitue un des arguments en faveur
du caractère monomoléculaire des derniers stades de la désamination nitreuse et
explique pourquoi de telles transpositions accompagnent souvent cette réaction.
Des recherches récentes (Cram, Hughes, Ingold, Winstein)
sur les conséquences stériques et sur la cinétique de ces transpositions ont
montré que les ions carbonium formés intermédiairement peuvent appartenir à
deux types limites dans le premier, la charge positive se trouve concentrée
essentiellement sur un seul atome de carbone; dans le second, cette charge est
répartie sur deux atomes de carbone contigus, lesquels sont reliés
simultanément par des valences partielles au radical migrateur R.
Cette dualité dans la nature des ions carbonium entraîne
une dualité correspondante dans le mécanisme des transpositions envisagées.
Dans certains cas, le départ du substituant X (X = ![]() dans le cas
particulier de la désamination nitreuse) précède la migration possible du
radical R (R = H, alcoyle ou aryle) ; l'ion carbonium (A) ainsi formé peut soit
réagir avec un des constituants Y du milieu réactionnel pour fournir un produit
de substitution "normale" sans migration du radical R, soit se
transformer en un ion carbonium (B) isomère, par migration du radical R, lequel
ion fournit des produits transposés (mécanisme "A").
dans le cas
particulier de la désamination nitreuse) précède la migration possible du
radical R (R = H, alcoyle ou aryle) ; l'ion carbonium (A) ainsi formé peut soit
réagir avec un des constituants Y du milieu réactionnel pour fournir un produit
de substitution "normale" sans migration du radical R, soit se
transformer en un ion carbonium (B) isomère, par migration du radical R, lequel
ion fournit des produits transposés (mécanisme "A").
Dans d'autres cas, le départ du substituant X est
accompagné d'un déplacement du radical R ; l'ion carbonium (C) formé peut
ensuite réagir avec un constituant Y du milieu réactionnel soit sur C , avec
formation d'un produit "normal", soit sur C, avec formation d'un
produit transposé (mécanisme "C") :

Telles sont, très brièvement esquissées, les théories
actuelles sur le mécanisme de la désamination nitreuse et des transpositions
rétropinacolique et pinacolique. Ces théories sont basées sur un ensemble de
faits extrêmement vaste ; cependant, aussi bien que les théories elles-mêmes,
ces faits présentent certaines lacunes et ce sont ces lacunes que nous nous
sommes efforcé de combler dans une certaine mesure dans le présent travail.
Exposé
d'ensemble
Le but de ce travail était de mettre en évidence et
d'étudier, en fonction du milieu réactionnel et de la structure du composé
envisagé, l'influence des conditions expérimentales sur la nature des composés
obtenus lors de la désamination nitreuse des a-amino-alcools. Nous avons ainsi été amené à étudier aussi bien
le mécanisme de la désamination nitreuse proprement dite que celui des
transpositions (rétropinacolique et pinacolique) qui accompagnent cette
réaction. D'autre part, nous avons généralisé certaines constatations
expérimentales nouvelles que nous avons faites au cours de ce travail.
Désamination nitreuse.- Les a-amino-alcools
sont, parmi tous les composés aminés, ceux qui ont été le plus étudiés au point
de vue de leur désamination ; en effet, celle-ci est susceptible d'apporter dos
renseignements d'un grand intérêt sur les aptitudes migratrices des différents
radicaux, et un grand nombre de travaux ont été publiés à ce sujet, surtout entre
1920 et 1940 (Bettzieche, McKenzie, Tiffeneau).
Mais ce point de vue a conduit les auteurs à préciser
surtout la nature des produits de la transposition qui accompagne en général la
désamination, sans se préoccuper de l'influence possible du milieu réactionnel
sur l'évolution de la réaction. Malgré les recherches qui avaient mis en
évidence l'influence des conditions expérimentales lors de la désamination
nitreuse des aminés simples, il semblait que, dans le cas des a-amino-alcools, la nature des produits
obtenus était entièrement prédéterminée par la seule structure de
l'amino-alcool envisagé. Il était admis, par exemple, que le formation d'un
produit de substitution (glycol) au lieu d'un produit transposé (aldéhyde ou
cétone), ou accompagnant celui-ci, ne pouvait avoir lieu que lorsque les
aptitudes migratrices des radicaux susceptibles de migrer étaient
particulièrement faibles.
Cependant, il y avait tout lieu de supposer que les
derniers stades de la désamination nitreuse de certains a-amino-alcools comportent, comme dans le
cas des aminés simples, la formation intermédiaire d'un ion carbonium (A).
Ainsi que l'a signalé Mlle Tchoubar en 1946, une telle interprétation laissait
prévoir qu'un changement dans les conditions expérimentales habituelles
entraînerait un changement correspondant dans la composition du produit de la
réaction. En particulier la présence, dans le milieu réactionnel, d'une forte
concentration en ions Y- (Y = Cl ouCH3-COO,
par exemple) devait être susceptible d'entraîner l'apparition, parmi les
produits habituels de la réaction [glycol (II) et cétone (III)], d'un
mono-ester (I, Y = Cl ou CH3COO) du glycol (II) par la réaction entre ces ions
et l'ion carbonium (A) :
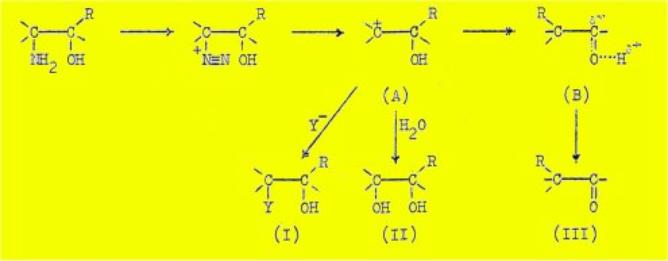
La formation d'un tel mono-ester serait en tous points
analogue à la formation des composés R-Y (Y = Cl ou CH3COO) dans le cas dos
aminés simples.
Les différentes recherches que nous avons effectuées sur
la désamination d'a-amino-alcools
de types variés ont conduit à des résultats qui confirment la prévision de Mlle
Tchoubar : les conditions expérimentales exercent une influence
considérable sur la nature des produits obtenus lors de la désamination
nitreuse des a-amino-alcools.
Cependant, nous avons constaté que ces résultats ne cadrent pas tous avec le
mécanisme monomoléculaire communément admis, et nous avons été amené à conclure
que la désamination peut également s'effectuer dans des conditions appropriées,
par d'autres mécanismes qui ne comportent pas le passage par un ion carbonium.
Nous avons, par la suite, confirmé et généralisé cette conclusion en étudiant
la désamination de deux amines simples, la néopentylamine et 1'a-phényléthylamine optiquement active.
Transpositions rétropinacolique et pinacolique.- Dans le cadre de ce travail sur la
désamination nitreuse, nous avons étudié, du point de vue théorique, le
mécanisme des transpositions qui accompagnent souvent cette réaction. Il existe
en effet des contradictions, déjà signalées par Tiffeneau, dans
l'interprétation actuellement admise de certains aspects des transpositions
rétropinacolique et pinacolique, notamment en ce qui concerne les aptitudes
migratrices relatives des différents radicaux aliphatiques et aromatiques d’une
part, et de l'hydrogène d'autre part.
De nombreux travaux ont été effectués (Tiffeneau) sur des
réactions transpositrices où il y a possibilité de choix entre deux ou
plusieurs radicaux (R, R' = H, alcoyle ou aryle) susceptibles de migrer :
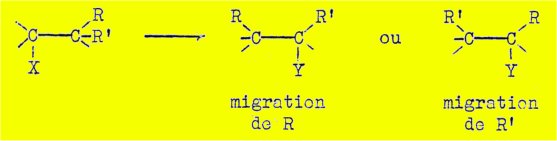
Ces travaux montrent que les radicaux aryle et alcoyle
sont très différents en ce qui concerne les rapports entre leurs aptitudes
migratrices et leur pouvoir donneur d'électrons (capacité affinitaire). Tandis
que pour les radicaux aryle, les aptitudes migratrices et les capacités
affinitaires vont souvent de pair, c'est-à-dire que ceux dont les capacités
affinitaires sont les plus fortes présentent généralement des aptitudes
migratrices plus marquées, il n'en est plus de même pour les radicaux alcoyle
et l'hydrogène, dont les aptitudes migratrices sont en général d'autant plus
faibles que les capacités affinitaires sont plus fortes.
Ces règles ne sont pas absolues et comportent un certain
nombre d'exceptions : les aptitudes migratrices des différents radicaux ne sont
pas des propriétés intrinsèques mais dépendant de la structure de la molécule
dans son ensemble (Tiffeneau).
Cependant, malgré ces exceptions, les règles énoncées
sont suffisamment générales pour permettre d'envisager entre les deux groupes
de radicaux (aryle et alcoyle) des différences profondes dont la nature restât
à élucider. En effet, il n'existait, au moment où nous avons entrepris le
présent travail, aucune explication valable ni de ces différences, ni de
certaines des exceptions observées, notamment en ce qui concerne les aptitudes
migratrices de l'hydrogène.
Nous avons ainsi été amené à rassembler et à confronter
les différents résultats obtenus lors de la désamination des composés aminés et
à envisager d'un point de vue nouveau les différents facteurs dont dépendent
les aptitudes migratrices des radicaux et la nature des produits obtenus lors
de réactions susceptibles de conduire à des transpositions de ce type. Cette
étude théorique nous a permis de proposer une explication rationnelle des
anomalies apparentes observées antérieurement.
Etudes connexes.- Les travaux que nous avons effectués sur la synthèse et la
désamination des a-amino-alcools
nous ont amené à faire un certain nombre de constatations expérimentales nouvelles
qu'il nous a paru intéressant d'approfondir et de généraliser. Ces différentes
recherches concernent : (l°) la synthèse d'a-amino-acides du type (R)2CNH2-COOH ; (2°) les réductions
sélectives au moyen de l'hydrure d'aluminium et de lithium; et (3°) l'oxydation
de certains époxydes et cétones par l'acide périodique.
Conclusions
Dans ce qui suit, nous nous efforcerons de dégager les
principales conclusions expérimentales et théoriques auxquelles nous ont
conduit les résultats obtenus au cours du présent travail. Certains de ces
résultats et conclusions ont déjà été sommairement exposés dans des notes et
communications préliminaires (Felkin, 1948, 1948a, l948b, 1950, l950a, 1951,
1951b, 1952, 1953).
Conclusions expérimentales.- Les conclusions expérimentales de ce
travail concernent d'une part la préparation et la désamination des composés
aminés que nous avons étudiés, et d'autre part les études expérimentales
connexes que nous avons entreprises.
(1°) Préparation des amino-alcools - Les a-amino-alcools que nous avons préparés
appartiennent à deux types : R-CHNH2-COH(R’)2 et (R’)2CNH-CH2OH.
Ceux du premier type, R-CHNH2-COH(R’)2 , ont été préparés
par la voie habituelle en faisant agir un organo-magnésien R’MgX sur le
chlorhydrate d’amino-ester R-CHNH2-COOR". Tous (R = H, R’ = C6H5 ; R
= C6H5, R’ = C2H5, CH2C6H5, C6H5) sauf un (R = H, R’ = C7H15) ont déjà été
décrits.
Par contre, lorsque nous avons entrepris ce travail, il
n’existait aucune méthode générale de préparation des amino-alcools du second
type, (R)2 CNH2-CH2Oh, dont un seul (R = CH3) avait été décrit. Après
différentes tentatives infructueuses nous avons élaboré une méthode permettant
d’obtenir un de ces composés (R = CH2C6H5). Elle consiste en 1a réduction au
moyen de l’hydrure d’aluminium et de lithium, suivie d’hydrolyse, de
l’acétamide-ester (R’)2 C(NHCOCH3)-COOR’.
Nous décrivons également différentes tentatives de
synthèse d’un a-amino-alcool
éthylénique R-CH=CHNH2-COH(R')2 dont nous voulions étudier la destination. Ces
tentatives n'ont pas abouti.
(2°) Influence des conditions expérimentales et de la
structure du composé initial sur la nature des produits de la domination nitreuse
des amine-alcools.- L’étude de la désamination nitreuse de différents
amino-alcools, dans des conditions variables, a confiné la prévision de Mlle
Tchoubar concernant 1’influence du milieu réactionnel sur la nature des
produits obtenus. En effet dans tous les cas que nous étudiés, nous avons
montré qu’un changement approprié des conditions expérimentales habituelles
entraîne une modification sensible, et parfois même radicale, dans la
composition du produit de la réaction.
Une expérience a été effectuée en présence d’alcool et
d’acide acétique ; elle a conduit à la formation, à côté du glycol et de la
cétone normalement attendus, d’une certaine proportion, déterminée par dosage,
de mono-acétine et de mono-éther du glycol.
Les autres expériences ont été effectuées en présence
d’acide chlorhydrique, soit en milieu aqueux (eau, ou eau + dioxane), soit en
milieu anhydre (dioxane).
L’acide chlorhydrique a été choisi car les chlorhydrines
obtenues sont facilement dosables dans le produit de la réaction ; par contre,
le dosage des autres constituants de ce produit (glycol et cétone) a présenté
des difficultés inattendues que nous n'avons que partiellement surmontées
Dans certains cas, les produits de la réaction
(chlorhydrine, glycol, cétone) ont été isolés à l'état pur et caractérisés.
Tous les a-amino-alcools
dont nous avons étudié la désamination en présence d'acide chlorhydrique ont
fourni une certaine proportion de chlorhydrine correspondante. La proportion de
chlorhydrine formée dépend d'une part des conditions expérimentales et d'autre
part de la structure de 1'amino-alcool étudié.
En ce qui concerne les conditions expérimentales, la
formation de chlorhydrine est favorisée par le présence d'une forte
concentration en acide chlorhydrique et, encore plus, par l'absence d'eau. La
proportion de chlorhydrine formée est plus forte dans un milieu anhydre
contenant une faible concentration d’acide chlorhydrique que dans un milieu
aqueux contenant une forte concentration de cet acide.
En ce qui concerne la structure de l'amino-alcool, la
formation de chlorhydrine est favorisée
par la présence d'un radical à fortes capacités affinitaires (phényle) sur
l'atome de carbone initialement porteur de l'azote, et par l'absence d'un tel
radical sur l'atome de carbone porteur de l'hydroxyle.
Lorsque les différentes conditions favorables (milieu
anhydre, forte concentration en acide chlorhydrique, présence d'un radical
phényle sur l'atome de carbone qui est le siège de la substitution) sont
réunies, la formation de chlorhydrine devient pratiquement exclusive.
L’influence des conditions expérimentales sur la
formation concomitante de glycol et de cétone a également été étudiée. En
milieu aqueux, lorsque la concentration en acide chlorhydrique augmente, la
chlorhydrine obtenue se forme surtout aux dépens de la cétone; au contraire,
pour une même concentration en acide chlorhydrique, lorsque la concentration en
eau diminue, la chlorhydrine se forme aux dépens aussi bien du glycol que de la
cétone.
Il résulte de cette étude que la composition du produit
obtenu lors de la désamination nitreuse d’un a-amino-alcool n’est pas entièrement prédéterminée par la
structure de celui-ci, comme on le croyait jusque présent, et qu’il est
possible de la faire varier d'une façon prévisible en modifiant les conditions
opératoires.
(3°) Influence des conditions expérimentales sur la
nature des produits de la désamination nitreuse de la néopentylamine. - II
est connu que la désamination de la néopentylamine dans les conditions
habituelles (milieu aqueux) conduit exclusivement à des produits transposés
(alcool amylique tertiaire et amylènes). Nous avons préparé cette amine par
réduction de l'amide correspondant au moyen de l'hydrure d'aluminium et de
lithium, et nous avons montré que sa désamination au moyen du chlorure de
nitrosyle en milieu anhydre (dioxane) conduit, à côté des produits transposés,
à la formation d'une faible proportion de chlorure de néopentyle.
(4°) Influence des conditions expérimentales sur les
conséquences stériques de la désamination nitreuse.- Nous avons étudié,
dans différentes conditions, la désamination nitreuse de 1'a-phényléthylamine optiquement active,
avec obtention du chlorure correspondant. L'amine a été préparée par la voie
habituelle à partir de l’acétophénone, et dédoublée (partiellement) au moyen
d'acide tartrique.
Nous avons montré que les conséquences stériques de la
désamination de l'a-phényléthylamine
dépendent étroitement des conditions expérimentale.
En milieu aqueux, la configuration du chlorure obtenu
n'est que faiblement influencée par la concentration du milieu en acide
chlorhydrique ; la réaction entraîne toujours une racémisation presque totale
(avec parfois une très faible inversion de l'ordre de 1 à 3 %).
En milieu dioxanique anhydre, la réaction effectuée avec
du chlorure de nitrosyle conduit à une conservation partielle (environ 40 %) de
la configuration spatiale ; comme en milieu aqueux, cette conséquence stérique
n'est que faiblement influencée par la concentration du milieu en acide
chlorhydrique.
Enfin, la désamination de 1 'a-phényléthylamine dans la pyridine
anhydre au moyen du chlorure de nitrosyle conduit à une inversion partielle
(environ 10 %) de la configuration spatiale.
(5°) Synthèse d'a-amino-acides du type (R)2 CNH2-COOH.- La recherche d'une méthode de
préparation des a-amino-alcools
du type (R)2 CNH2-CH2OH nous a amené à étudier la synthèse des a-amino-acides correspondants. Nous avons
préparé des dérivés de ces composés par trois voies différentes.
En premier lieu, nous avons montré que la transposition
de Beckmann, appliquée aux b-oximino-esters
CH3-C(=NOH)-C(R)2-COOC2H5, conduit aux a-acétamido-esters (R)2 C(NHCOCH3)-COOC2H5, accompagnés de leurs
produits d'hydrolyse (R)2 C(NHCOCH3)-COOH et (R)2 CNH2-COOH. Cette réaction a
été effectuée avec deux oximino-esters (R = C2H5, C4H9) ; elle a échoué avec un
troisième (R = CH2C6H5).
Cette réaction constitue une nouvelle méthode d'obtention
de certains a-amino-acides. Il n'existe aucune
corrélation apparente entre l'aptitude des b-oximino-esters à se transposer et leur aptitude à se cycliser.
En effet, les deux oximino-esters (R = C2H5 et CH2C6H5) se sont cyclisés avec
une très grande facilité (formation d'isoxazolone) alors que l'oximino-estcr (R = C4H9) n’a pu être cyclisé.
En deuxième lieu, nous avons préparé l'acétamido-ester (R
= CH2C6H5), que nous n'avions pu obtenir par la transposition de Beckmann, par
action de l'acide azothydrique (Schmidt) sur le céto-ester
CH3-CO-C(R’)2-COOC2H5.
Enfin, nous avons préparé
l'amino-acide (R = C6H5) par hydrolyse, au moyen de la baryte,, de la
diphénylhydantoïne.
(6°) Réductions sélectives au moyen de l'hydrure
d'aluminium et de lithium.- Par^la synthèse d'un acétamido-alcool
(R)2C(NHCOCH3)-CH2OH (R = CH2C6H5) à partir de l'acétamido-ester correspondant
(R)2 C(NHCOCH3)-COOC2H5 au moyen de l'hydrure d'aluminium et de lithium
(réduction de la seule fonction ester), nous avons mis en évidence pour la
première fois la possibilité d'effectuer des réductions sélectives au moyen de
ce réactif.
Nous avons généralisé cette observation en montrant que
l’hydrure double permet souvent de réduire sélectivement une seule fonction
dans les composés comportant deux fonctions différentes, toutes deux
réductibles. C'est ainsi qu'il est possible, dans certaines conditions que nous
avons précisées, de réduire sélectivement la fonction cétone des
oximino-cétones et des cétones chlorées, et la fonction ester des
oximino-esters, des nitro-esters et des acylamido-esters.
Cette sélectivité dans l'action de l'hydrure d'aluminium
et de lithium est susceptible d'avoir un certain nombre d'applications en
chimie organique.
(7°) Oxydation de certains époxydes et cétones par
l'acide périodique.-
Les aléas que nous avons rencontrés lors de nos essais de dosage, au moyen de
l'acide périodique, des a-glycols
présents dans le mélange obtenu lors de la désamination de certains a-amino-alcools, nous ont amené à étudier
l'action, de ce réactif sur les autres constituants du mélange.
Nous avons pu montrer ainsi que l'acide périodique n'est
pas aussi spécifique qu'on ne l'avait cru jusqu'à présent, car il attaque
certains époxydes et les arylacétones du type Ar-CHR-CO-R'.
Conclusions théoriques.- Les conclusions théoriques de ce travail concernent d'une part
le mécanisme des derniers 'stades (R-N![]() N
N ![]() produits) de la
réaction de désamination nitreuse, et d'autre part le mécanisme des
transpositions du type rétropinacolique et pinacolique qui accompagnent très
souvent cette réaction.
produits) de la
réaction de désamination nitreuse, et d'autre part le mécanisme des
transpositions du type rétropinacolique et pinacolique qui accompagnent très
souvent cette réaction.
(l°) Mécanisme do la désamination nitreuse.-Les résultats obtenus dans le présent
travail montrent que la désamination nitreuse, comme beaucoup d'autres
réactions en chimie organique, peut se dérouler suivant au moins deux
mécanismes différents. Le mécanisme monomoléculaire (a), communément admis,
n'est en réalité pas le seul mécanisme suivant lequel peuvent s'effectuer les
derniers stades de cette réaction, car il existe, à côté de ce mécanisme, un
mécanisme intramoléculaire (b) de désamination, et vraisemblablement aussi, un
mécanisme bimoléculaire (c) :

Les conséquences aussi bien structurales (formation de
produits transposés ou non transposés) que stériques (inversion ou conservation
de la configuration, racémisation) de ces trois mécanismes sont différentes ;
elles ont permis de les distinguer.
Le mécanisme suivant lequel s'effectue la désamination nitreuse
dépend des conditions expérimentales choisies et de la structure du composé
étudié. L'analyse détaillée d'une part des résultats du présent travail et
d'autre part de ceux de certaines recherches antérieures, restées inexpliquées
jusqu'alors, ont permis de préciser et d'interpréter dans une certaine mesure
1'influence.de ces deux facteurs.
Le mécanisme monomoléculaire (a) comporte comme
conséquence stérique une racémisation importante accompagnée d'une inversion
partielle de la configuration spatiale ; il peut conduire à des transpositions.
La plupart des résultats obtenus antérieurement et les résultats obtenus dans
le présent travail, lors de la désamination des a-amino-alcools et de. l'a-phényléthylamine optiquement active,
montrent que la désamination de tous les composés aminés dans un milieu
contenant une très forte proportion d'eau se fait presque toujours par ce
mécanisme. Ce n'est que lorsque la structure du composé étudié entrave d'une
manière quelconque le mécanisme monomoléculaire que la réaction a lieu, même en
milieu aqueux par un autre mécanisme (intramoléculaire). S'il a été admis
jusqu'à maintenant que le mécanisme monomoléculaire était le seul mécanisme de
la désamination nitreuse, cela tient essentiellement au fait que le mécanisme
de cette réaction a presque toujours été étudié en milieu aqueux et avec des
composés dont la structure ne présentait aucune entrave à l'opération de ce
mécanisme.
Le mécanisme intramoléculaire (b) comporte comme
conséquence stérique la conservation de la configuration spatiale ; il conduit
toujours à des composés de substitution "normaux", sans
transposition. Ce mécanisme comporte la formation transitoire d'un composé
diazoïque covalent, en équilibre avec l'ion diazonium, et le passage par un
état de transition cyclique :
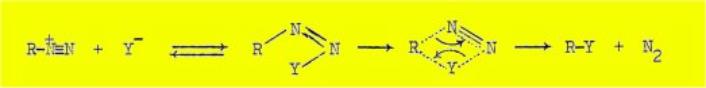
Les résultats obtenus dans le présent travail, lors de la
désamination des a-amino-alcools,
de la néopentylamine, et de 1'a-phényléthylamine
optiquement active ainsi que dans certaines recherches antérieures, montrent
que ce mécanisme est favorisé par des conditions qui déplacent l'équilibre
diazonium-diazoïque vers la forme diazoïque. Ces conditions sont d'une part un
milieu anhydre (dioxane, acide acétique), moins ionisant que l'eau, et d'autre
part, mais à un degré moindre, la présence dans ce milieu d'une forte
concentration en anion Y-. L'influence de la structure a été moins
nettement précisée; mais les résultats obtenus avec les a-amino-alcools indiquent que le
mécanisme intramoléculaire est favorisé par la présence d'un substituant à
forte capacité affinitaire (phényle) sur l'atome de carbone qui est le siège de
la substitution, et par l'absence d'un tel substituant sur l'atome de carbone
porteur du groupement hydroxyle ; d'autre part, ce mécanisme est favorisé par
tout facteur structural qui entrave le mécanisme monomoléculaire.
Le mécanisme bimoléculaire (c) comporte comme conséquence
stérique l'inversion totale de la configuration spatiale ; comme le mécanisme
intramoléculaire, il ne doit conduire qu'à des composés de substitution
"normaux », sans transposition. Les résultats obtenus dans le présent
travail lors de la désamination de l'a-phényléthylamine optiquement active au sein de la pyridine
indiquent que dans ces conditions cette réaction s'effectue partiellement
suivant le mécanisme bimoléculaire.
Cette pluralité de mécanismes lors de la désamination
nitreuse, mise en évidence pour la première fols dans ce travail, pose certains
problèmes théoriques et ouvre certaines perspectives, tant théoriques
qu'expérimentales, que nous avons brièvement esquissés.
(2°) Mécanisme des transpositions rétropinacolique et
pinacolique. - Nous avons examiné en détail les différents facteurs qui
déterminent le mécanisme ("A" ou "C") des transpositions de
ce type et la nature des produits auxquels elles conduisent (aptitudes
migratrices des radicaux R et R'). Cette étude a conduit à une conception unifiée
et cohérente de ces transpositions, qui semble tenir compte de tous les faits
expérimentaux actuellement connus.
Nous montrons que la relation entre les capacités
affinitaires et les aptitudes migratrices des différents radicaux aryle ou
alcoyle, et de l'hydrogène, dépend directement du mécanisme même suivant lequel
s'effectue la transposition.
Lorsque la transposition a lieu par le mécanisme
"A", la composition du produit de la réaction est déterminée par la
stabilité relative des ions (B et B’) auxquels l’ion initial (A) peut conduire
par transposition.
Or, l'ion le plus stable, dont dérivera le produit de la
réaction, est celui dont le carbone chargé positivement est porteur des
radicaux à plus fortes capacités affinitaires. Les radicaux qui ne migrent pas
sont donc ceux à capacités affinitaires les plus fortes ou, en d'autres termes,
le radical migrateur est celui ayant la plus faible capacité affinitaire.

Au contraire, lorsque la transposition a lieu par le
mécanisme "C", la composition, du produit de la réaction est
déterminée par la stabilité relative des ions (C) et (C’) ;
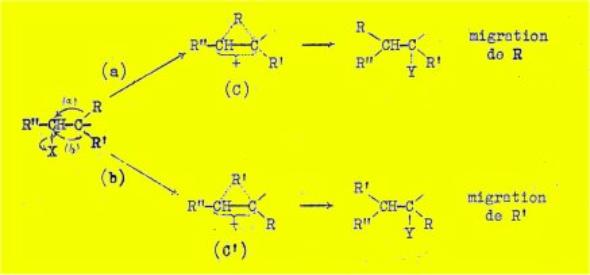
Or, l'ion le plus stable, dont dérivera le produit de la
réaction, est celui dans lequel le radical qui est relié simultanément aux deux
atomes de carbone, possède la plus forte capacité affinitaire. Le radical
migrateur est donc celui ayant la plus forte capacité affinitaire.
Le fait que les aptitudes migratrices et les capacités
affinitaires, des radicaux aryle vont généralement de pair, alors que c'est le
contraire chez les radicaux alcoyle et l'hydrogène, est dû simplement à ce que
la migration des radicaux aryle (à fortes capacités affinitaires) a
généralement lieu par le mécanisme "C", alors que celle des radicaux
alcoyle et de l'hydrogène (à faibles capacités affinitaires) a généralement
lieu par le mécanisme "A". De même, le fait que la migration des
radicaux aryle 1’emporte en général sur celle des radicaux alcoyle et de
l'hydrogène est dû à ce que lorsqu'il y a possibilité de choix, la réaction se
fait de préférence par le mécanisme "C". L'étude des transpositions
de composés cyclaniques (extensions et diminutions de cycle) montre que les
chaînons de cycles carbonés se comportent en général comme les radicaux aryle.
Cependant, ce ne sont là que des règles générales qui
présentent un certain nombre d'exceptions : on sait que les aptitudes
migratrices ne sont pas des propriétés intrinsèques des radicaux mais dépendent
de la structure de la molécule envisagée dans son ensemble. Cela revient à dire
que le mécanisme suivant lequel s'effectuent les transpositions
rétropinacolique et pinacolique dépend non seulement de la nature du radical
migrateur mais également d'autres facteurs. Ces facteurs sont : la nature des
radicaux qui ne migrent pas ; la nature du groupement éliminé X ; la
configuration spatiale de la molécule ; et la nature des conditions
expérimentales.
Le premier de ces facteurs (la nature des radicaux qui ne
migrent pas) paraît être le plus important et explique en particulier les
anomalies apparentes observées en ce qui concerne l'aptitude migratrice de
l'hydrogène. Les radicaux aryle à fortes capacités affinitaires, migrent
généralement par le mécanisme "C" de préférence à l'hydrogène dont
les capacités affinitaires sont très faibles et dont la migration s'effectue
par le mécanisme "A" ; cependant, dans certains cas, c'est le
contraire qui se passe : l'hydrogène migre de préférence à un radical aryle. On
a tenté (Tiffeneau), et on tente encore (Ingold), d'expliquer ce fait en
admettant que la réaction s’effectue par un mécanisme vinylique et ne comporte
donc pas une véritable migration de l'hydrogène. En réalité, cependant, cette
"anomalie" est due à l'influence, sur le mécanisme même de la
transposition, des radicaux fixés sur l'atome de carbone porteur (à l’origine)
du substituant X qui est éliminé. Lorsque ces radicaux sont à faibles capacités
affinitaires, l'ion (C) est plus stable que l'ion (A) , la réaction a lieu
« normalement" et la migration d'un aryle, par le mécanisme
"C", a lieu de préférence à celle d'un hydrogène, par le. mécanisme
« A". Au contraire, lorsque ces radicaux possèdent de fortes
capacités affinitaires, ils contribuent à stabiliser l’ion (A).à tel point que
c'est cet ion qui se forme lors du départ du substituant X et non l'ion (C) ;
dans ces conditions, la réaction a lieu par le mécanisme « A » et
c’est l'hydrogène, à faibles, capacités affinitaires, qui migre de préférence à
un radical aryle.
La conception esquissée ci-dessus a permis d'interpréter
un certain nombre de faits restés inexpliqués jusqu'à présent, par la simple
étude comparée des aptitudes migratrices et des capacités affinitaires des
différents radicaux, elle permet également de déterminer dans de très nombreux
cas le mécanisme ("A" ou "C ») suivant lequel s’effectuent
les transpositions du type rétropinacolique et pinacolique ; lorsque les
aptitudes migratrices et les capacités affinitaires vont de pair, la réaction a
lieu par le mécanisme "C"; dans le cas contraire, la réaction a lieu
par le mécanisme "A".
Cette méthode d'investigation du mécanisme de ces
réactions est beaucoup plus générale que celles qui ont été employées
antérieurement (étude de la cinétique et des conséquences stériques de la
transposition), et qui n'ont pu être appliquées qu'à un nombre de cas
relativement restreint. Là où il existe des points de comparaison, les
résultats obtenus par ces différentes méthodes concordent.
Mots clefs : alcoyle / aliphatique / aluminium / amine /
amino-acide / amino-alcool / aryle / carbone / carbonium / cétone / chlorhydrine /
chlorhydrique / chlorure / cinétique / concentration / condition / configuration /
conséquence / désamination / dioxane / dosage / exception / facteur / fonction / glycol /
interprétation / intramoléculaire / inversion / lithium / mécanisme / méthode /
monomoléculaire / néopentylamine / oximino-ester / phényléthylamine / pinacolique /
racémisation / réaction / résultat / rétropinacolique / structure / synthèse / tchoubar /
tiffeneau / transposition / felkin / bauer