Titres et travaux scientifiques
de
Irène Felkin
I.
MECANISMES REACTIONNELS
ET STEREOCHIMIE
1.
Mécanisme
et stéréochimie de la transposition cétolique
Bien que la transposition
cétolique ait été découverte il y a plus de 50 ans (Favorsky), elle n'a pas été
aussi bien étudiée que la plupart des autres transpositions.
Les travaux les plus intéressants
sur cette réaction ont été faits jusqu'à présent sur des cétols stéroïdes.
La transposition cétolique
consiste en l'isomérisation d'un cétol en un autre :

elle peut avoir lieu chez les
cétols secondaires (R’ = H) ou tertiaires.
Par son mécanisme elle
s'apparente aux transpositions au type benzilique, provoquées par les agents
alcalins, et à celles provoquées par les agents acides, telles que la
transposition pinacolique.
En effet la transposition
cétolique peut être provoquée par des agents acides et alcalins.
Les principaux résultats de
l'étude de la transposition cétolique, que .j'ai effectuée soit seule, soit en
collaboration avec Mlle B. Tchoubar et Mmes Le Ny et Skrobek, sont les suivants
:
1°- La transposition peut
être effectuée, aussi lien en série aromatique qu'en série aliphatique et
cyclanique, au moyen du tertiobutylate d'aluminium, qui est un catalyseur plus
général de cette réaction que les acides protoniques ou les alcalis.
Le mécanisme ci-dessous rend
compte des faits connus actuellement [réversitilité, formation d'alcool
t-C4H9OH, stéréosélectivité] :

2°- Dans la série
cyclanique, la transposition cétolique peut avoir lieu, soit avec extension,
soit avec diminution de cycle; le radical qui migre dans ce dernier cas est un
chaînon du cycle carboné :

Les cétols cyclopentaniques
nécessaires à cette étude ont été préparés par une méthode que j’ai élaborée (voir
paragraphes).
La réaction d'extension de cycle
constitue une bonne méthode de préparation de cétols cyclohexaniques à
carbonyle intracyclique.
3°- L’étude des dimères des
cétols a permis de découvrir une transposition nouvelle : sous l'influence des
agents acides, et en particulier de l'acide formique, le dimère (VI)
s'isomérise, avec migration du radical phényle, en ester du cétol monomère :

Cette réaction constitue le premier exemple
de transposition d’un a-cétol tertiaire
en acide isomère.
4°- Certains faits observés
par différents auteurs lors de l'étude des cétols stéroïdes n'avaient pas reçu
d'explication. Ainsi par exemple, il a été établi que l'isomérisation du cétol
de structure partielle (I), qui se fait avec extension du cycle D, fournit un
mélange de deux cétols D-homo isomères, résultant de la migration des carbones
C16 et C13 sur le carbone C20 :

Le cétol (II) se forme en
quantité prépondérante, alors qu'on s'attendrait normalement à la formation
prédominante de l'isomère (III), résultant de la migration du carbone C13, le
plus substitué et donc le plus nucléophile.
J'ai expliqué ce résultat, puis
j'ai confirmé expérimentalement cette explication, de la façon suivante. L'état
de transition, conduisant au cétol cyclohexanique (II) possède la conformation
chaise, alors que l'état de transition correspondant à la formation du cétol
cyclohexanique (III) possède la conformation bateau.
Le cétol (II) se forme donc plus
rapide ment que son isomère (III).

Une étude cinétique de cette réaction a confirmé ce point de
vue. La même hypothèse a été émise quelques années plus tard par des auteurs
américains (Mendier, Taub et Firestone, Experientia, 1959, 15, 237),
mais ils n'ont pas encore fait d'étude cinétique de la réaction.
5°- Au cours de l'étude des
cétols stéroïdes, j'ai montré pour la première fois que les D-homo cétols du
type (II) et (III) peuvent se transformer l'un dans l'autre dans différentes
conditions.
Une étude détaillée du mécanisme
de cette transformation a permis de montrer qu'elle a lieu non pas directement,
avec migration du méthyle de C17 à C17a et vice versa, mais indirectement, par
l'intermédiaire du cétol (I) avec contraction, puis extension du cycle D,
suivant le schéma ci-dessous :

En 1959, les mêmes auteurs américains ont démontré
indépendamment ce mécanisme.
2. Relation entre structure et stabilités des composés
isomères
II a été maintes fois constaté que de nombreux composés
carbonylés (cétols et cétones) possédant dans leur molécule un groupement
CO-C6H5 s'isomérisent en composés carbonylés correspondants, possédant un
groupement CO-CH3.
Ces faits n'ont pas reçu d’explication jusqu'à présent,
aussi ai-je entrepris une étude systématique de l'isomérisation de tels
composés dans l'espoir d'élucider la relation entre leur structure et leur
stabilité.
Avec Mlle Verrier et Mr Colard j'ai étudié la position de
l'équilibre suivant, à différentes températures :

Les résultats obtenus montrent
que la différence de stabilité entre ces deux isomères correspond surtout à une
variation d'enthalpie du système en équilibre. La variation d'entropie est très
faible (entre 50° et 120°).
La différence de stabilité ne
peut être attribuée uniquement à la présence ou à l'absence de liaison
hydrogène intramoléculaire dans les deux isomères.
En effet, les deux cétols sont
presque entièrement chélatés (spectres IR). D’ailleurs nous avons montré que la
position d'équilibre chez les cétones correspondantes, possédant la même
structure carbonée et qui ne peuvent pas se chélater, est également en faveur
du composé non conjugué :

Une étude détaillée des
propriétés physiques et chimiques des isomères de ce type est susceptible de
mettre en évidence des facteurs de stabilisation autres que la chélation,
l'hyperconjugaison et la conjugaison, ce qui permettrait d'expliquer les faits
observés.
Le rôle de plusieurs facteurs
susceptibles de stabiliser les cétols non conjugués a été mis en évidence; le
gain énergétique dû à chacun de ces facteurs est inférieur à 1'énergie de
conjugaison, mais le gain total est suffisamment important pour rendre, dans de
nombreux cas, les cétols non conjugués plus stables que leurs isomères
conjugués.
Un facteur de stabilisation des
cétols non conjugués, qui n'a pas été reconnu jusqu'à présent, a été mis en
évidence; la stabilisation provient de l'interaction du carbonyle avec le
groupement aromatique porté par le carbone en a du carbonyle.
Le gain énergétique dû à cette
interaction, qui se manifeste dans les spectres ultraviolets vers 290 mm, a été
estimé approximativement à 2 kcal.
3. Transposition des a-glycols
et de leurs mono-éthers
La déshydratation des a-glycols
par les agents acides a été beaucoup étudiée par l'école Tiffeneau. Il ressort,
entre autres, de ces travaux, que la déshydratation des glycols
secondaires-tertiaires (I) conduit aux aldéhydes (II), avec migration du
radical R, lorsque ce radical est aromatique.
Par contre, dans le cas où ce
radical est un alcoyle à faible poids moléculaire, la déshydratation, effectuée
dans les mêmes conditions, conduit aux cétones (III) avec migration de
l'hydrogène:

En étudiant faction des agents
acides sur les mono-éthers de glycols de formule générale (IV), j'ai montré que
les aptitudes migratrices relatives des radicaux aliphatiques et de l'hydrogène
sont liées d'une façon étroite à la structure de la molécule dans son ensemble.
En effet, les radicaux
aliphatiques à faible poids moléculaire, même le méthyle, migrent aussi bien ou
mieux que l'hydrogène lorsque le radical R" du groupe alcoxy est un
alcoyle à poids moléculaire élevé (de C4H9 à C7H15 :

La présence dans la molécule d'un
radical R" cyclanique tel que le cyclohexyle ou le menthyle favorise au
contraire la migration de l'hydrogène (formation de cétone).
Le mécanisme de cette réaction
peut être représenté schématiquement de la façon suivante :

Le point essentiel de ce
mécanisme est que la migration du radical R accompagne ou précède le départ du
groupe R". Il est clair, en effet, que si le groupe R" s'éliminait
avant la migration du radical R, il ne saurait favoriser cette migration.
Les résultats de ce travail sur
les a-glycols et leurs mono-éthers ont fait l'objet de ma thèse
et ont été communiqués au Colloque international sur les réarrangements
moléculaires et l'inversion de Walden.
4. Désamination nitreuse des
aminés et amino-alcools cyclaniques
La désamination nitreuse des
composés aminés cyclaniques peut conduire, lorsque la fonction aminé est
juxtacyclique, à une extension de cycle carboné.
C’est ainsi que la désamination
des amines cyclaniques (I, R=H) conduit aux alcools (II) (réaction de Demjanov)
et la désamination des amino-alcools (IV, R=H) conduit aux cétones (V)
(réaction de Tiffeneau-Tchoubar) :

l'extension de cycle est
généralement accompagnée d'une réaction compétitive conduisant, respectivement,
à des alcools (III) ou à des a-glycols (VI) de même structure carbonée que le composé
aminé de départ.
La proportion relative des
produits transposés et non transposés obtenus dépend de la stabilité relative
des deux ions (A) et (B) qui se forment intermédiairement dans ces réactions :

L'extension de cycle ne peut
avoir lieu d'une façon appréciable que si l'ion transposé (B) est plus stable
que l'ion initial (A).
J'ai étudié, en collaboration
avec Mlle B. Tchoubar et Mme Y. Gault, la désamination de plusieurs aminés (I)
ou amino-alcools (IV) (n = 4 ou 5, R = alcoyle à chaîne linéaire ou ramifiée,
cyclohexyle, aryle).
La plupart des amino-alcools
nécessaires à cette étude ont été préparés par une méthode que j'ai mise au
point (voir paragraphe 8).
Cette étude a permis de dégager
et d'interpréter l'influence des différents facteurs structuraux sur la
composition du produit de la désamination.
Ces facteurs sont les
suivants :
1°- Le nombre de chaînons du
cycle carboné : l'extension de cycle se
fait plus aisément chez les composés cyclopentaniques (n = 4) que chez les
composés cyclohexaniques (n = 5), toutes choses égales d'ailleurs.
2°- La nature du composé aminé : l'extension de cycle est plus générale chez les
amino-alcools que chez les aminés.
3°- La nature du radical R : l'extension de cycle est défavorisée ou même empêchée
totalement par les radicaux R aromatiques.
4°- L'étude des amino-alcools
(VII), comportant un radical ramifié, a montré que dans ce cas la migration
d'un chaînon de cycle est en compétition avec une seconde transposition,
conduisant à la formation d'un b-glycol (VIII) par l'intermédiaire
de l'ion (C) :

La formation de b-glycols
lors de la désamination des a-amino-alcools n'avait encore jamais été observée.
5. Préparation et stéréochimie des b-arylsérines
L'élaboration d'une synthèse du
chloramphénicol a amené H. Feikin, Z. Welvart et moi-même à étudier les b-arylsérines,
matières premières nécessaires à cette synthèse.
Il s'agissait essentiellement de
trouver un moyen d'accès commode à une b-arylserine, ou à un dérivé de ce
composé, possédant la configuration thréo, comme le chloramphénicol.
Nous avons élaboré des méthodes
de synthèse stéréosélectives des b-arylsérines et nous avons ensuite
étudié les mécanismes par lesquels se forment ces composés pour tenter
d'expliquer la stéréosélectivité observée.
Une partie des résultats obtenus
a été communiquée au Colloque sur les réactions d'hydroxyalcoylation et au Colloque International sur
l'hydroxycarbonylation.
1°- Synthèse de l’érythro b-phénylsérine
La réduction catalytique sur
platine de l'oximino benzoylacétate d'éthyle conduit presque exclusivement à 1'érythro
p-phénylsérinate d'éthyle :

2°- Synthèse du thréo b-p-nitrophénylsérinate
d'éthyle
Le chloramphénicol possède dans
sa molécule un groupement nitro qu'il est avantageux d'introduire aussi près
que possible du début de la synthèse.
Nous avons cherché à préparer un
dérivé de la nitrophénylsérine par condensation de l'aldéhyde p-nitro-benzoïque
avec l'ester du glycocolle.
En effectuant cette condensation
en présence d'un excès de glycocollate d'éthyle nous avons pu orienter la
condensation vers la formation exclusive d'un dérivé (II) de configuration
thréo :

3°- Mécanisme de formation des
dérivés érythro et thréo de la b-p-nitrophénylsérine
Le premier stade de la réaction
entre l'aldéhyde p-nitrobenzoïque et l'ester du glycocolle est la formation
d'un composé d'addition (III) (Ar = NO2C6H4) :

Ce stade est commun à la
formation des deux composés érythro (I) et thréo (II).
Le mécanisme de la formation du
dérivé érythro est le suivant :

Cette condensation, comme nous
l'avons montré, est catalysée par les agents alcalins.
Le mécanisme de formation du
dérivé thréo (II) n'a pas été entièrement élucidé. Un certain nombre de faits
que nous avons établis suggèrent un mécanisme bimoléculaire :

4°- Transhydroxyalcoylations dans
la série des b-arylsérines
Lorsque l'on traite le composé
érythro (I) par l'ester méthylique du glycocolle en excès on observe la
réaction suivante (Ar = NO2C6H4) :

Cette réaction constitue une
transhydroxyalcoylation du glycocollate de méthyle par le composé (I).
L'étude du mécanisme de la
transhydroxyalcoylation effectuée avec le composé (I) optiquement actif a
montré qu'il y a racémisation presque totale au cours de la réaction.
Il y a donc rupture de la liaison
entre les deux centres d'asymétrie de la molécule (Ca et Cb) au
cours de cette réaction :

La rupture de la liaison Ca-Cb se fait
par le mécanisme inverse de celui qui figure à la page précédente.
L'aldéhyde aromatique libéré
réagit ensuite avec l'ester du glycocolle en excès comme précédemment. Des
réactions du même type dans la série des phénylsérines ont été étudiées
récemment en Hongrie par A. Hajos dont les résultats confirment les nôtres.
5°- Configuration absolue des
quatre b-p-nitrophénylsérinates d'éthyle
L'amino-ester thréo (II) a été
dédoublé en antipodes optiques au moyen de l'acide lactique.
La configuration absolue des
énantiomorphes thréo et érythro a été établie. L'énantiomorphe thréo dextrogyre
a été transformé en chloramphénicol dont la configuration est connue.
L'amino-ester érythro a été
dédoublé par une méthode connue et chacun des énantiomorphes a été transformé,
par une suite d'opérations comportant une inversion sur le carbone porteur de
l'hydroxyle, en amino-ester thréo de configuration précédemment établie :

6°- Stéréochimie des réactions
d'addition sur le carbonyle
J'ai étudié, avec H. Felkin, la
stéréochimie de l'addition des dérivés organo-magnésiens sur les cétones.
L'étude de l'action du chlorure d'a-phényléthyl magnésium sur
l'acétaldéhyde a montré que la réaction conduit surtout à 1'érythro phényl-3
butanol-2 (I) (>80%). Elle est donc très stéréosélective :

La formation prédominante du
dérivé érythro (I) est due à ce que les interactions stériques entre les
groupements les plus volumineux des molécules réagissantes sont moindres dans
l'état de transition qui mène au dérivé érythro que dans celui qui mène au
dérivé thréo (II). L'alcool érythro se forme donc plus facilement, ou, ce qui
revient au même, plus rapidement que l'alcool thréo.
A l'occasion de cette étude, nous
avons montré également qu'il est possible de séparer les alcools
diastéréoisomères (I) et (II) par chromatographie en phase gazeuse.
II. METHODES DE SYNTHESE
7.- Synthèse du
chloramphénicol (chloromycétine)
Après, plusieurs tâtonnements, la
synthèse du chloramphénicol entreprise, sous l'impulsion de Mlle B. Tchoubar, a
été réalisée par H. Feikin, Z. Welvart et moi-même à partir du thréo b-p-nitro-phénylsérinate
d'éthyle (I).
Le problème de la synthèse du
chloramphénicol à partir de ce composé se ramène à la réduction sélective du
groupement ester, sans réduction du groupe nitro :

Nous nous sommes proposés
d'effectuer cette réduction sélective du groupe ester au moyen de l'hydrure
d'aluminium et de lithium. Cependant, de nombreuses expériences préliminaires,
effectuées sur le phénylsérinate d'éthyle (IV) qui ne possède pas de groupe
nitro, ont montré qu'il était sans espoir de tenter de réduire sélectivement le
nitro-ester (I) par ce réactif.
Nous avons montré alors que le
blocage des fonctions aminé et alcool du phénylsérinate d'éthyle (IV) par création
d'un noyau oxazoline (VI, R = CH3 ou C6H5) constitue un moyen de protection
efficace de la molécule vis-à-vis de l'hydrure double.

Le même blocage s'est avéré
efficace dans le cas du nitro-ester (I) ; le groupe ester de l'oxazoline (V)
qui en dérive peut être réduit sélectivement par l'hydrure d'aluminium et de
lithium.
Notre but principal, la réduction
sélective du groupe ester, étant atteint, nous avons mis au point une synthèse
en quatre étapes du chloramphénicol (III) en employant l'imino dichloracétate
d'éthyle pour former le noyau oxazoline :

8. Synthèse des a-cétols et a-amino-alcools
à fonction alcool tertiaire
Aprèse avoir essayé, puis rejeté,
un certain nombre de voies d'accès
possibles aux a-cétols, j'ai montré que les cyanhydrines des cétones
peuvent, dans certaines conditions, constituer une matière première pour la
synthèse de ces composés.
Les cyanhydrines des cétones se
dissocient généralement sous l'action des organo-magnésiens ; le blocage de la
fonction hydroxyle des cyanhydrines au moyen du dihydropyranne permet d'obtenir
la réaction d'addition normale du réactif de Grignard sur la fonction nitrile
sans dissociation de la molécule ;
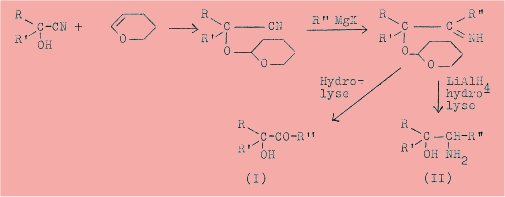
L'action de l'hydrure d'aluminium
et de lithium sur les cétimines formées intermédiairement, suivie d'hydrolyse
de la fonction acétal, permet d'accéder facilement aux amino-alcools (II).
Cette voie de synthèse s'est avérée
très générale; elle a été beaucoup employée dans notre laboratoire et a permis
de préparer un grand nombre de composés difficilement accessibles par ailleurs.
Les cyanhydrines de la cyclopentanone, de la cyclohexanone, de la
cycloheptanone, de l'acétone et de la méthyl éthyl cétone ont pu être
transformées aisément de cette façon en cétols (I) ou amino-alcools (II)
comportant des radicaux R" aliphatiques linéaires ou ramifiés, cyclaniques
ou aromatiques.
Dernièrement cette même méthode a
été appliquée par des auteurs italiens aux cyanhydrines de cétones stéroïdes.
Ils ont obtenu des cétols stéroïdes avec un bon rendement (P. de Ruggieri et C,
Ferrari, J. Amer. Chem. Soc., 1959, 81, 5725).
Une variante de la même méthode a
été employée également avec succès par Nazarov, Ahrem et Kamernitzky (Zh.
Obshch. Khim. 1958, 1805).
Mots clefs : acide / aminé / amino-alcool / aromatique / carbone / cétolique /
cétol / cétone / chloramphénicol / composé / configuration / cyanhydrine / cyclanique / cycle / dérivé /
désamination / érythro / ester / éthyle / étude / fonction / glycocolle / hydrure / isomère / mécanisme /
méthode / migration / molécule / radical / réaction / stéréochimie / stéroïde / structure / synthèse /
thréo / felkin