Thèse présentée à la faculté des sciences de l’Université de Paris
pour obtenir le grade de Docteur ès sciences physiques
par
Pierre RIVAILLE
LA TRANSESTERIFICATION DES PHENYL-PHOSPHATES CYCLIQUES DE GLYCOSIDES EN MILIEU ALCALIN
soutenue le 02 avril 1962
devant la commission d’examen :
Le Breton, présidente
Lederer, M. Julia, examinateurs
INTRODUCTION
Depuis plusieurs années déjà, biochimistes et chimistes se sont penchés sur l'étude des esters de l'acide phosphorique, les uns pour élucider leur rôle comme métabolites intermédiaires, les autres pour déterminer leur structure et étudier leurs réactions.
Dès 1935, Bailly et Gaumé ont établi que l'hydrolyse alcaline des méthyl glycérophosphate a et b donnait un mélange de glycérophosphates a et b sans formation de méthyl phosphate.

Par la suite Bally s’est aperçu qu'en milieu acide les formes a et b de l'acide glycérophosphorique s'isomérisaient.
VERKADE formula l'hypothèse que cette isomérisation se produisait par l'intermédiaire d'une cylisation, et CHARGAFF utilisant du phosphore marqué la confirma, et démontra que ce réarrangement était intramoléculaire.

En 1948, BAER et KATES concluaient à la suite de leurs travaux, que la migration du groupe phosphate dans la molécule de méthylglycérophosphate venant du catabolisme des céphalines du cerveau et des lécithines, s'opérait par l'intermédiaire d'un phosphate cyclique.
Déjà en 1942, FONO avait entrevu l'importance des groupes hydroxyles voisins du phosphate sur l'instabilité des diesters phosphoriques et il avançait l'hypothèse de la formation d'un cycle intermédiaire sur ces hydroxyles pendant les réactions d'hydrolyse des esters phosphoriques.
CARTER et COHN, après avoir hydrolysé l'acide ribonucléique de levure ont séparé deux acides adényliques isomères a et b et par la suite, les trois autres nucléotides ont été séparés par paires d'isomères.
CARTER a suggéré alors que l'acide adénylique a était l’adénosine 2 phosphate et le b l'adénosine 3 phosphate.
BROWN et TODD ont démontré que ces deux isomères restaient stables en milieu alcalin, mais étaient interconvertibles en milieu acide.
Ils ont postulé que cette conversion s'effectuait par l'intermédiaire d'un cycle et proposé alors pour un mécanisme précis de l'hydrolyse du méthyl glycéro phosphate observée précédemment le passage par la forme d'un triester neutre.
Des travaux effectués dès 1922 avaient montré que si les acides ribonucléiques étaient dégradés par des bases en mono-nucléotides, les acides désoxyribonucléiques où le constituant glucidique ne possède pas deux groupes hydroxyles voisins restaient stables en milieu alcalin.
L'ensemble de ces recherches ont permis à TODD et à ses collaborateurs d'appliquer la théorie de cyclisation intermédiaire dans la dégradation alcaline des polynucléotides .
La liaison phosphorique entre les chaînes de nucléosides serait scindée entre l'hydroxyle en 5 et le phosphate, puis il se produirait une cyclisation sur l'hydroxyle en 2 suivie d'une hydrolyse donnant naissance aux nucléosides -2- et -3-phosphate.
Cette théorie se trouva confirmée par les travaux de MARKHAM et SMITH qui isolèrent les nucléosides 2, 3 phosphate à partir de l'acide ribonucléique et les identifièrent à ceux synthétisés par BROWN, MARGRATH et TODD.
En 1954, LIPKIN suivit le processus de l'hydrolyse alcaline des acides ribonucléiques, soit dans un milieu enrichi en 18O, soit avec du méthoxyde de sodium et, à la suite des travaux de BLUMENTHAL et HERBERT et de JORDAN avança que le mécanisme de réaction s'effectuait par les étapes suivantes.


Ces hypothèses sur le passage par des cycles intermédiaires dans l'hydrolyse des acides ribonucléiques amenèrent d'autres chercheurs à étudier la formation de ces cycles phosphorylés sur les nucléosides.
En particulier DEKKER et KHORANA en traitant les acides adényliques a et b par le dicyclohexycarbododiimide (DCC) avaient retrouvé de l'adénosine-2, 3-phosphate cyclique qui par hydrolyse acide ou basique redonnait les produits de départ.

Tous les cycles phosphorylés dont nous venons de parler, qu'ils furent synthétisés ou supposés comme intermédiaires de réactions, comportaient cinq chaînons.
Le premier cycle à six chaînons fut le méthyl-a-D-glucoside-4, 6-phosphate, préparé par BADDILEY, BUCHANAN et SZABÔ en traitant le méthyl-a- D-glucoside-4, 6-phényl phosphate par hydrogénolyse sur platine d'Adams.
On avait déjà soupçonné l'existence d'un tel cycle à la suite des travaux de LEVENE et RAYMOND qui avaient vainement tenté de synthétiser le xylose-3-phosphate sans passer par l'intermédiaire d'un cycle, par phosphorylation directe du 1,2 isopropylidène-5-O-acylxylofuranose.
Après l'élimination des groupes protecteurs en milieu acide, ils ne trouvaient que du xylose-5-phosphate ; dans ce milieu, le phosphate migrait de l'hydroxyle en 3 à celui en 5 et cette migration devait se produire par l'intermédiaire d'une cyclisation, non plus sur un hydroxyle secondaire voisin du phosphate, mais sur l'hydroxyle primaire qui est assez éloigné, et le cycle ainsi formé comportait six atomes.
En 1957, MOFFATT et KHORANA synthétisèrent le xylose-3-phosphate par hydrolyse alcaline du 1,2 isopropylidène xylofuranose-3, 5-phosphate cyclique (A).
Ils obtenaient cet autre cycle à six chaînons par action du dichlorophénylphosphate sur le 1, 2-isopropylidène xylofuranose (B).
Pour hydrolyser le groupe phényle du triester phosphorique et ouvrir le cycle en un mélange de 1,2 isopropylidène xylofuranose 3 et 5 phosphate (C et D) KHORANA et MOFFATT ont soumis le 1,2 isopropylidène xylofuranose 3,5-phényl phosphate à un traitement alcalin puis ils libéraient le phosphate de sucre du groupe isopropylidène par une hydrolyse acide très ménagée (E et F).
La séparation des deux phosphates de sucre isomères s'effectue ensuite par chromatographie sur résine.

Au fur et à mesure que les recherches progressaient dans le domaine des esters phosphoriques cycliques, d'autres problèmes se levaient. Ainsi dans un travail en collaboration avec TENER, WRIGTH et KHORANA, MOFFATT étudie les propriétés des cycles à cinq ou six chaînons et avance que dans les composés de formule générale :

si l'hydroxyle est dans une position favorable, il se produit en milieu acide ou alcalin une transestérification conduisant à la formation d'un composé cyclique :
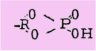
Il constate de plus que les monoesters phosphoriques des composés polyhydroxylés par le dicyclohexylcarbodiimide fournissent invariablement un diester cyclique à cinq chaînons même si la formation d'un cycle à six chaînons, est stériquement possible.
Le premier cycle à sept chaînons, d'ailleurs non fermé sur un glucide : le butane diol 1,4 phosphate cyclique fut synthétisé par MOFFATT et al. qui ont par la suite comparé les stabilités des cycles phosphorylés suivant le nombre d'atomes qui les composent.
Dans cette étude il ressort que les cycles à cinq chaînons sont beaucoup plus sensibles que ceux à six chaînons envers les acides et les bases.
P. SZABO et L. SZABO synthétisèrent un cycle à sept chaînons bâti sur les hydroxyles en 3 et 6 du glucofuranose et apportèrent à la théorie de MOFFATT l'argument supplémentaire que les cycles à cinq chaînons se forment de préférence même s'il est stériquement possible d'en créer à six ou sept chaînons.
Ces mêmes auteurs étudiant l'hydrolyse alcaline des cycles à six chaînons tels les méthyl-a-D-glucoside et -b-D-galactoside-4, 6-phosphate cyclique ont observé que contrairement à ce qui se passe dans le cas de l'acide panthoténique -2.4-phosphate cyclique où l'on ne retrouve qu'un seul monoester, le 4 phosphate, ces phosphates de sucre s'hydrolysaient en un mélange de 4 et 6 phosphate sans que les hydroxyles en 2 et 3 interviennent dans la réaction.
La cinétique de l'hydrolyse des esters phosphoriques a été étudiée dès 1942; BAILLY et DESJOBERT, les premiers montrèrent que l'hydrolyse du glycérol phosphate et de l'éthyl phosphate était maximale à un pH d'environ 4.
BUTCHER et WESTHEIMER obtinrent la même résultat avec le -2-méthoxypropyl-1-méthyl éthyl phosphate et constatèrent que cet ester optiquement actif était hydrolysé avec une rétention totale de configuration qui indiquait la fission de la liaison P-O.
OLDHAM avec de l'eau marquée en 18O hydrolysa le méthyl phosphate à pH 4 et retrouva du méthanol et du phosphate inorganique totalement enrichi en 18O il annonça alors que seule la liaison P-O était clivée.
Des expériences faites sur le b-D-glucose 1 phosphate révélèrent que si à des pH compris entre 1 et 4 l'hydrolyse se faisait par fission de la liaison C-O, à pH 6 ou. 7, elle avait lieu par rupture de la liaison P-O.
VERNON et ses collaborateurs rapportent que dans l'hydrolyse de l'aglycone des glucosides, les deux mécanismes avaient été observés, et qu'il était difficile d'établir exactement le processus de la réaction.
L'hydrolyse alcaline des triesters de l'acide phosphorique a été fort. peu étudiée.
Les premiers travaux sur ce sujet ont été faits par BARNARD, VERNON et THAIN sur les triéthyl, triméthyl et triphényl phosphate qui, en solution dans du dioxanne aqueux, s'hydrolysent au contact d'une base par rupture de la liaison P-O.
En milieu acide, les résultats obtenus sont moins nets et ne permettent pas d'affirmer le sens de la réaction.
Les derniers travaux parus sur l'hydrolyse des esters phosphoriques sont ceux de HAAK et WESTHEIMER.
On savait depuis longtemps que l'hydrolyse des esters phosphoriques à cinq chaînons, à cause des tensions internes du cycle, était beaucoup plus rapide que celle des esters à chaînes ouvertes correspondants.
Ainsi par exemple, la chaleur d'hydrolyse du méthyl éthylène phosphate est de 7 à 9 K cal/mol, supérieure à celle du diméthyl hydroxyéthyl phosphate.
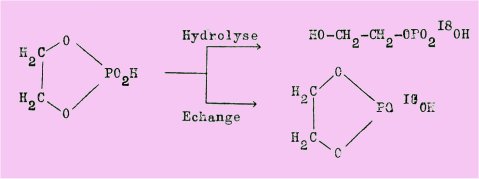
HAAK et WESTHEIMER conduisirent leurs travaux sur l'hydrolyse de l'éthanediol 1, 2 phosphate en solution acide ou basique enrichie en 18O, et observèrent que quelles que soient les conditions, la rupture de la liaison P-O se produit environ 300 fois plus vite que celle de la liaison C-O, et que cette dernière ne se fait à peu près pas ; mais si dans un milieu alcalin, il n'y a pas d'échange entre l'oxygène du solvant et un oxygène du phosphate ce phénomène a lieu en milieu acide.
Les auteurs symbolisèrent ces réactions par les équations suivantes :
Des travaux que nous avons cités, il ressort que si les monoesters et les diesters phosphoriques des glucides ont été très étudiés, l'hydrolyse des triesters est fort peu connue.
KHORANA seul précise que le 1, 2 isopropylidène xylofuranose 3,5-(phényl phosphate) est hydrolyse en milieu alcalin avec perte du groupe phényle.
Dans le travail que nous présentons, nous avons étudié les cas des méthyl et phényl glucosides et phényl galactoside 4, 6 (phényl phosphate) et constaté que quand un hydroxyle voisin du phosphate est libre ce qui n'était pas le cas dans le 1, 2 isopropylidène xylofuranose 3 5-(phényl phosphate) synthétisé par KHORANA, ces triesters subissent en milieu alcalin, une transestérification intramoléculaire aboutissant à la formation de glycosides 3 et 4 phosphate.
Nous avons démontré que la nature du groupe aglycone influe sur les proportions des monoesters formés pendant l'hydrolyse alcaline des glycosides 4, 6 phosphate cyclique. Cette influence du groupe aglycone se manifeste aussi sur l'acidité de ces esters phosphoriques et sur les réactivités des hydroxyles libres sur la molécule du glycoside.
Nous avons synthétisé tous les monoesters de l'acide phosphorique des phényl glucosides et galactosides et nous avons mis au point des méthodes pour les analyser sur résines échangeuses d'ions.
Nos observations pendant ces analyses ou ces synthèses semblent être des points de départ intéressants pour des travaux ultérieurs, surtout que COLLINS vient de mettre en évidence dans des produits naturels l'existence de triesters phosphoriques.
RESUME ET CONCLUSIONS GENERALES
La migration du radical phosphoryle des mono- et diesters de l'acide phosphorique est un phénomène connu.
Elle a lieu à la suite d'une cyclisation chaque fois que la molécule qu'estérifie l'acide phosphorique contient un ou plusieurs groupes hydroxyles situés dans une position stérique favorable à la formation des cycles.
Cependant on ignore le comportement des triesters de l'acide phosphorique qui sont pourtant des intermédiaires dans la synthèse des diesters.
Lors de la préparation de diesters (les méthyl- et phényl glucoside 4, 6-phosphates) à partir de triesters les -4, 6-(phényl phosphates) correspondants, nous avons remarqué la formation simultanée des monoesters et prouvé qu'ils ne provenait pas de l'hydrolyse alcaline du méthyl-a-D-glucoside -4, 6-(phényl phosphate) et du phényl-b-D-galactoside-4, 6-(phényl phosphate). Le produit principal de la réac-tion séparé par chromatographie des monoesters formés simultanément a été identifié comme étant toujours le glucoside 4, 6-(hydrogène phosphate).
L'analyse des mono-esters par des techniques chromatographiques et des titrages périodiques a révélé que dans les trois cas examinés ce mélange contenait une plus grande quantité de glycoside-3-phosphate que de glycoside-4-phosphate.
La présence de glycoside-3-phosphate en quantité prépondérante est remarquable puisque l’hydroxyle en 3 était libre dans la molécule originale.
De plus comme le glycoside-6-phosphate est absent dans le mélange des monoesters si par un changement de technique l'hydrolyse ou par la nature de la molécule hydrolysée la fraction des mono phosphates devenait le produit principal de la réaction, les conclusions tirées de l'examen des produits d'hydrolyse pour établir la structure originale de la molécule risqueraient d'être fausses.
Si les observations de COLLINS, selon lesquelles des triesters de l'acide phosphorique existeraient dans des produits naturels se trouveraient confirmées, ces résultats pourraient avoir un certain intérêt biochimique.
Pour établir la structure des mono phosphates formés pendant cette transestérification tous les mono phosphates des phényl-b-D-glucopyranoside et galactopyranoside, à savoir les 2, 3, 4 et 6-phosphate s ont été synthétisés .
Nous avons trouvé que l'oxychlorure de phosphore, dont l'emploi avait été plus ou moins abandonné tant il est difficile de purifier le produit phosphorylé, est le meilleur agent phosphorylant dans la synthèse des monoesters de l'acide phosphorique, chaque fois que le support glucidique peut être rendu hydrophobe par un choix judicieux des groupes protecteurs des hydroxyles.
En préparant les intermédiaires pour la synthèse de certains monophosphates, nous avons pu démontrer que les réactivités des différents hydroxyles des glycosides varient suivant la nature du groupe aglycone.
Ainsi, sur le méthyl 4, 6-benzylidène b-D-glucoside l'attaque du chlorure de benzoyiese fait de préférence sur l'hydroxyle en 2, mais c'est sur celui en 3 qu'elle a lieu dans le cas des phényl 4,6-benzylidène bD-glucoside et -galactoside.
Pour les synthèses des phényl glucoside- et galactoside-3- phosphates, malgré de nombreux essais, nous ne sommes pas parvenu à obtenir un dérivé des phényl glycosides dont seule la fonction hydroxyle en 3 restât libre.
Nous avons donc préparé les 4, 6-benzylidène phényl glycoside 2, 3-(phényl phosphates) : leur hydrolyse donnait un mélange de phényl glycoside-2 et 3-phosphates dont le 3-phosphate, prédominant, fut isolé.
Cette suite de réactions confirmait nos résultats antérieurs sur la réactivité plus grande du groupe hydroxyle en 3 des phényl gluco- et galactopyranosides.
Le groupe aglycone des glycosides phosphorylés n'a pas d'influence sur la composition de la fraction des mono phosphates formés par transestérifieation lors de l'hydrolyse des 4, 6(phényl phosphates) des glycosides ; en revanche, il modifie les proportions relatives des 4- et 6-phosphates obtenus par l'hydrolyse des 4, 6-(hydrogène phosphate).
Ainsi, après l'hydrolyse alcaline des méthyl-a-D-glucoside- et méthyl-b-L-galactoside-4, 6-phosphates cycliques, on retrouve 4 à 5 fois plus des méthyl glycoside 4- phosphates que des méthyl glycoside 6-phosphates, tandis que le même traitement des phényl-b-D-glucoside- et-galactoside-4, 6-phosphates cycliques ne donne que 2 à 3 fois plus de glycoside 4-phosphate que de glycoside 6-phosphate.
Nous avons identifié les différents mono phosphates formés sur résine échangeuse d'ions après avoir étalonné les colonnes avec les monophosphates authentiques des glycosides.
Pendant cette analyse, nous avons observé que le groupe aglycone des glycosides avait une grande influence sur l'acidité des phosphates de sucre.
Les phosphates des phényl glycosides sont des acides plus forts que ceux des méthyl glycosides et cette différence est accrue par les complexes que forment les sucres avec l'ion borate.
Nous avons vainement tenté d'élaborer une synthèse du méthyl-a-D-glucoside-3, 6 phosphate, que nous avons considéré tout d'abord comme un intermédiaire possible pendant l'hydrolyse alcaline du méthyl-a-D-glucoside 4, 6-phénylphosphate.
Ce composé contiendrait un cycle phosphorylé à sept chaînons, bâti sur le noyau pyranoïde de l'hexose.
Or sur des modèles moléculaires les tensions à exercer sur la molécule pour former ce cycle sont très fortes, ce qui rend peu probable sa formation et explique nos résultats négatifs.
Pour ces expériences nous avons synthétisé le méthyl 2, 4 di-O-benzyl-a-D-glucopyranoside et nous avons essayé de le phosphoryler soit avec le dichlorophényl phosphate ou par réaction sur le sucre du diphénylchlorophosphate suivie d'un traitement alcalin.
Dans les deux cas, le seul produit que nous pu isoler était le méthyl 2, 4-di-O-benzyl-a-D-gluccside 6-(phényl phosphate).
Ceci nous a permis de démontrer que l'hydrolyse du phényle des phénylphosphates de glucide ne peut s'opérer que si les 2 autres fonction de l'acide phosphorique sont estérifiées ; les diphényl-phosphates de glucide se transforment en diesters de l'acide phosphorique cyclisés sur la molécule du sucre, si la cyclisation n'est pas possible seul un des deux groupes phényles est éliminé.
MOTS CLEFS : acide / aglycone / alcalin / chainon / cycle / cyclique / cyclisation / diester / ester / formation / galactoside / glucide / glucoside / glycoside / hydrolyse / hydroxyle / intermédiaire / isomères / isopropylidène / liaison / mélange / méthyl / molécule / monoester / nucléoside / phényl / phosphate / phosphorique / réaction / résultat / ribonucléique / structure / sucre / synthèse / triester / xylofuranose / xylose